FDA Advisors Positive on Sentinel Cerebral Protection Device in TAVR
There was no formal vote following today’s discussions, but most panelists agree that potential benefits of protection outweigh any risks.

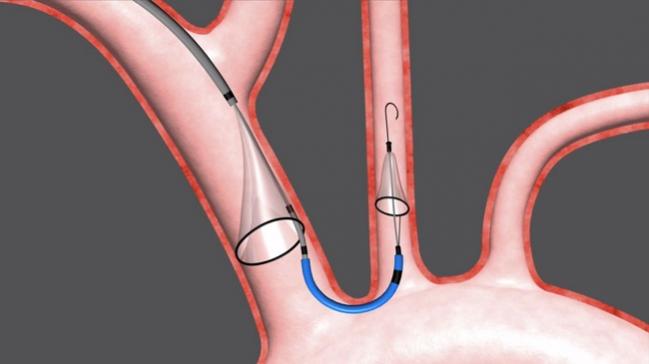
Advisors to the US Food and Drug Administration (FDA) agreed today that the Sentinel Cerebral Protection System, used during transcatheter aortic valve procedures, is safe and likely effective, paving the way for the approval of the first such device in the United States. Most, despite concerns about the lack of clear clinical benefit, said they themselves would want to have the device in place if they underwent such a procedure.
While panel members took no formal vote—this first-in-class device follows a “de novo” approval pathway—their closing comments indicated that they believe the totality of the data suggests that it may be of benefit and they would want it on the market. That’s despite the fact that the pivotal SENTINEL trial met its primary safety endpoint but did not meet its primary efficacy endpoint of reduction in brain lesion volume on MRI.
“If we were voting, I would vote in favor of approval,” David Naftel, PhD (University of Alabama at Birmingham), observed, echoing the remarks of others on the panel.
“Intuitively, it seems like a very good thing to me that these [bits of] debris are taken out of the circulation pretty effectively by this filter and I think that’s a good thing,” concluded Jeffrey S. Borer, MD (SUNY Downstate Medical Center, New York, NY). "And even though I really can’t interpret what it means, I intuitively believe that the images that were done look better when a filter was used than when a filter wasn’t used. . . . I’m troubled that I don’t have a clear position that I can take. However, if I had to, if I was being forced to [choose], I’d rather have this thing in than not have it in, if I had to have a TAVR."
One of the dissenting voices, Kevin M. Duff, MD (University of Utah, Salt Lake City), said he had hoped to see positive results today, but didn’t. “I hope that the work continues in this, but I don’t feel as strongly about it (as my fellow panel members).”
Magnus Ohman, MD (Duke University, Durham, NC), likewise, said he felt the panel was lost somewhere “between science and emotion,” but that he ultimately didn’t feel the trial was persuasive enough to convince him. “I’m favorably disposed, but I’m highly uncertain.”
The FDA does not always follow the advice of its advisory panels, but today’s largely unanimous support will be viewed positively by market-watchers. The device is already approved in Europe and elsewhere.
Standing Guard
Members of the agency’s Circulatory System Devices Panel spent much of the day wrestling with the results of SENTINEL and other studies. In particular, panelists grappled with the use of the occurrence of DW-MRI lesions as the surrogate endpoint, with most agreeing that this likely was not the appropriate choice, simply because these brain lesions are not known to correlate with clinical stroke or other neurocognitive defects. That said, most agreed that the lesions are likely “real” and may have an important neurological impact long term.
The percutaneous device is delivered prior to a TAVR procedure via the right radial artery and consists of two cap-shaped filters. One filter is delivered to the brachiocephalic artery, the other to the left common carotid artery. Debris dislodged during the procedure—typically calcification, thrombus, or valve tissue—is captured in the filters and removed with the device at the end of the procedure.
The left posterior inferior cerebellar artery is left unprotected by the filter, but according to the sponsor, this supplies just 2% of brain tissue.
As previously reported by TCTMD, SENTINEL randomized 363 patients undergoing TAVR at 18 US and European centers, 2:1, to cerebral protection or no cerebral protection. Debris was found in 99% of cases; however, MACCE rates in the cerebral protection group were noninferior to the performance goal and not statistically different from patients treated without the protection device (7.3% vs 9.9%; P = 0.41). Likewise, new lesion volume was numerically lower but not statistically different between the protection and no protection groups (102.8 vs 178.0 mm3; P = 0.33). After adjusting for baseline lesion volume and valve type, however, there was a significant reduction in new lesion volume in protected territories (P = 0.02) with embolic protection.
An internal review of the evidence conducted by FDA reviewers prior to today’s meeting concluded that physicians and patients have a “strong desire for cerebral protection and there are currently no approved alternatives. The Sentinel System provides clinical benefit and the risks can be mitigated.”
Indeed, panel members today found it hard to ignore the debris—some of it sizeable—that the the filters caught in most of the patients in the study. This is despite the quip by one panel member that "just because there is stuff in the strainer, that doesn't mean you approve the strainer."
Summing up the day's discussion, chair Richard Page, MD (University of Wisconsin, Madison), observed that the panel consensus was that the device was safe, that the study was flawed (but perhaps could not have been done better and won't be done again), and that the debris capture is compelling.
"I think we’re all in agreement," he said, "that we’d rather have that coming out of our body than going into our brain."
Photo credit: Adapted from Claret Medical animation: http://www.claretmedical.com/technology/us-sentinel-cps/

Shelley Wood was the Editor-in-Chief of TCTMD and the Editorial Director at the Cardiovascular Research Foundation (CRF) from October 2015…
Read Full Bio
Comments