FDA Advisory Panel Gives Thumbs Up to EVAR System
The INCRAFT stent graft system for treating infrarenal AAA overcame concerns about higher numbers of certain adverse events.

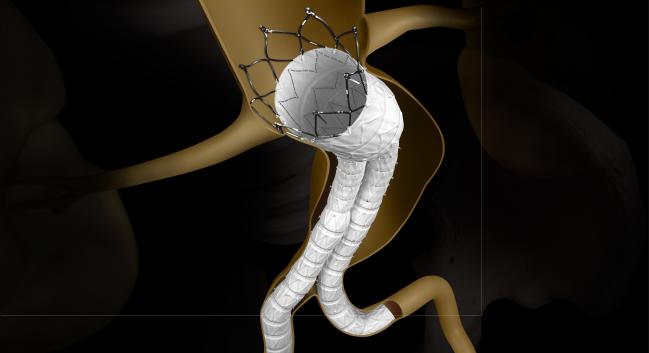
An advisory panel of the US Food and Drug Administration (FDA) has endorsed the premarket approval application for the INCRAFT stent graft system (Cordis) for treating infrarenal abdominal aortic aneurysms (AAAs) despite concerns about increases in selected adverse events
On Tuesday, members of the Circulatory System Devices panel of the FDA’s Medical Devices Advisory Committee voted in support of the system’s safety (11-4) and effectiveness (14-0, with one abstention) for use in patients who meet criteria listed in a relatively broad proposed indication. The majority of panelists (11-4) thought the benefits outweighed the risks.
The daylong vetting included a look at data from the INSPIRATION trial, a prospective, multicenter, single-arm study of the INCRAFT system, which was approved in Europe in 2014 and is now available in 39 countries, according to a press release from Cardinal Health, Cordis’ parent company. The study met its primary endpoints for safety (major adverse events through 30 days) and effectiveness (successful aneurysm treatment through 1 year), and there were no aneurysm ruptures through 4 years of follow-up.
Cordis filed a premarket approval application with the FDA in February 2015, but the agency issued “Not Approvable” letters on three separate occasions, requesting more information regarding certain adverse events that were detected in greater numbers in INSPIRATION than in trials of other endovascular aneurysm repair (EVAR) devices.
The stated purpose of Tuesday’s meeting, according to FDA briefing documents, was to “discuss how the high rate of observation of transrenal stent fractures, patency-related events, aneurysm expansion associated with Type II endoleaks, and secondary interventions occurring through 5-years of follow-up impact the safety and effectiveness, as well as the benefit/risk profile, of the device system.”
The panel was unanimous or near unanimous in seeing the system as one that fills an unmet clinical need, according to chairperson Richard Page, MD (University of Wisconsin School of Medicine and Public Health, Madison).
“The issue for discussion was whether it would be appropriate for all patients or whether the indication or the eventual use would be more narrow for, specifically, a population with tortuous or smaller-diameter iliacs,” he told TCTMD. “And there remains some concern—at least, the majority expressed some concern—about the findings from the clinical trial. But at the same time, the overall outcomes of these patients were excellent in terms of the most important endpoint, and that would be death or morbidity related to abdominal aortic aneurysm rupture, which was not an issue in this trial.”
Page noted that even though there are several EVAR systems currently available in the United States, the INCRAFT system has some unique characteristics that would expand the numbers of patients with infrarenal AAA who could be treated. Page pointed to the low profile of the system and maneuverability in tortuous or smaller-diameter iliac vessels, whereas the FDA cited a higher proposed suprarenal neck angulation limit compared with other devices.
Though “the concerns raised by the FDA I think are valid concerns,” panelists were provided with some reassurance by the fact that there were no aneurysm ruptures in the INSPIRATION trial, Page said. Some members of the panel also proposed that the higher numbers of certain adverse events could be due to enhanced surveillance or differences in patient selection rather than device-specific issues.
Page did not cast an official vote because he was chair of the panel, but he said he would have voted “No” on the question whether the benefits of the INCRAFT system outweigh the risks for use in patients who meet the criteria in the proposed indication. That was not necessarily a vote against eventual approval, however.
Panelists who voted “Yes” to that question were voting for the system to be approved for the broad proposed indication, with the expectation that vascular surgeons would choose the right device for each individual patient, Page said. “I believe those who voted ‘No’ were not voting that the device should not be approved, but were voting on the specific indication that was provided, and would have rather seen FDA and the sponsor [propose] a more narrow indication for the device because of the concerns that were raised.”
Recapping comments he made after the vote, Page recalled: “I said that this actually meets an unmet need, but I would have voted against it because I felt that the broad indication was not appropriate.”
Considering the higher numbers of some adverse events, “perhaps for an uncomplicated case that would be amenable to any device out there, [the INCRAFT system] would not be the first choice,” Page said. “On the other hand, there was, I think, full agreement that for certain patients this would be the device of choice.”
The panel also weighed in on a postapproval study proposed by Cordis, which would incorporate 150 patients from a European postapproval study and 150 new US patients, with follow-up lasting up to 5 years. Page said the panelists, who were not enthusiastic about the idea of combining European data with different levels of follow-up with the data from new patients, made various recommendations to develop the study with appropriate endpoints. They stressed, too, that enough women should be enrolled in order to get a better understanding of how the INCRAFT system works among those with smaller-sized iliac arteries.
Photo Credit: Cardinal Health

Todd Neale is the Associate News Editor for TCTMD and a Senior Medical Journalist. He got his start in journalism at …
Read Full BioSources
Cardinal Health. FDA advisory committee votes in favor of Cardinal Health’s INCRAFT AAA stent graft system for the endovascular treatment of infrarenal abdominal aortic aneurysms. Published on: June 12, 2018. Accessed on: June 14, 2018.
Comments